
|
|
|
|
| GENETIK |
Die Erhaltung genetischer Vielfalt (mit einem populationsgenetischen Modell) |
|
|
|
|

|
Das Hardy Weinberg Gesetz (Wikipedia) |
|
|
(1) VorwortGenetische Forschung kann auf verschiedenen Stufen betrieben werden, auf der molekularen Stufe, auf der zellulären Stufe, auf der Stufe des Individuums und auf der Stufe der Population. Jede der Betrachtungsweisen führt zur Entdeckung verschiedener Gesetzmäßigkeiten. Die Regeln im biochemischen Bereich werden auf populationsgenetischer Ebene nicht erforscht und nicht erkannt. Das gilt auch umgekehrt. Der Populations- bzw. Evolutionsgenetiker sollte aber alle Ebenen gut kennen. Er bedient sich aber vorwiegend – aber nicht ausschließlich – der klassischen Genetik. Die Bedeutung der Evolutionsgenetik ergibt sich aus ihren Querverbindungen mit anderen Disziplinen. Da Veränderungen am Genbestand einer Population als evolutive Prozesse aufgefasst werden müssen, ergibt sich, dass die Evolutionsgenetik einen wesentlichen Anteil an der Klärung der Fragen der Evolution hat. Dies gilt nicht nur für Pflanze und Tier, sondern auch für den Menschen. Auch menschliche Populationen haben einen gemeinsamen Genbesitz, der Völker und Nationen, ja die gesamte Menschheit verbindet. Pflanzen- und Tierzüchtung haben Erfahrungen über die Möglichkeiten und Grenzen einer künstlichen Selektion gesammelt, die zu einer gerichteten Veränderung des Genpools der Nutzpflanzen- und Nutztierpopulationen geführt haben. Die genetische Zusammensetzung einer Population wird sowohl durch Selektionsprozesse als auch durch Zufallsverteilungen und verschiedene Kombinationsprozesse der Erbfaktoren bestimmt. Einiges lässt sich durch mathematische Modelle darstellen, daher ist die experimentelle Populationsgenetik ein besonders interessanter Zweig der Evolutionsgenetik. Die moderne Evolutionsgenetik erlebte in den Siebzigerjahren einen enormen Aufschwung – vor allem in angelsächsischen Ländern. Nachdem das Interesse an der Gentechnik gegen Ende der Siebzigerjahre absolut dominierend geworden war, geriet die Evolutionsgenetik etwas in Vergessenheit und fristet seither ein wenig beachtetes Dasein. In Wahrheit kann die Evolutionsgenetik einen enormen Beitrag zur Evolutionsbiologie bringen. Dieser Aufsatz soll daran erinnern. (2) Die genetische VielfaltDer englische Biologe Charles Darwin hatte erkannt, dass die Selektion eine der wichtigsten Motoren der Evolution des Lebens ist. An dieser zentralen Theorie Darwins hat sich bis heute nichts geändert. Es ergab sich jedoch eine entscheidende Frage, die Darwin im 19. Jahrhundert – im Gegensatz zu heute - nicht beantworten konnte, denn Darwin kannte weder die Mendelschen Erbgesetze noch ihre evolutionsbiologischen Konsequenzen. Deren Kenntnis hätte einige der damals offenen Fragen beantwortet. Selektion kann es nur dort geschehen, wo es Vielfalt gibt. Warum aber gibt es überhaupt Vielfalt? Woher kommt sie? Warum ist sie so unübersehbar, obwohl erstaunlich viele Mutationen so genannte Letal- oder Subletalmutationen sind? Man muss kein Evolutionsgenetiker sein, um zu erkennen, welche genetische Vielfalt in biologischen Arten steckt. Nicht nur der Wolf zeigt einen verblüffenden genetischen Polymorphismus, die bei den vielen gezüchteten Hunderassen sichtbar wird. Wenn von genetischer Vielfalt die Rede ist, müssen einige Begriffe geklärt werden. Ein "Gen" wird in der Evolutionsgenetik meist als "Genlocus" bezeichnet. Liegt dieses Gen in zwei oder mehr verschiedenen Ausführungen vor (beispielsweise Blutgruppen beim Menschen), dann spricht man von Allelen.
Abb 1. Chromosom III von Drosophila melanogaster mit den durch Kreuzungsversuche gewonnen Positionen einiger Genloci. se und st sind sichtbare Mutanten (Augenfarbe) Idh (Isozitratdehydrogenase) und Pgm (Phosphoglucomutase) sind Enzymloci. Da es bei Idh und Pgm mehrere Allele gibt, die man nur mittels Elektrophorese identifizieren kann, spricht man von "Allozymen". Ly und D sind Drosophila-Flügelmutanten, die in einer Inversion (Chromosomenmutation) "gefangen" sind. Aus (4).
Wenn wir von Vielfalt (Polymorphismus) sprechen, sind einige zusätzliche Überlegungen wichtig. Unter dem genetischen Polymorphismus versteht man die Erscheinung, dass in einer Population einer Tier- oder Pflanzenart oder des Menschen mehrere verschiedene und unterscheidbare Formen nebeneinander vorkommen, deren Unterschiede erblich sind. Im einfachsten Fall ist das darauf rückführbar sein, dass von einem Gen(locus) mehrere verschiedene Allele in einer Population vorkommen. In vielen Fällen wird es sich um komplexe determinierte Eigenschaften handeln. Je nach der Wirkung des Gens oder des Genkomplexes wird sich das in einer Population so auswirken, dass Individuen verschiedener Färbung (Farbpolymorphismus), verschiedener Gestalt (morphologischer Polymorphismus) oder verschiedenen Verhaltens (Verhaltenspolymorphismus) parallel auftreten. In vielen Fällen lassen sich die verschiedenen Genotypen nicht ohne besondere Hilfsmittel voneinander unterscheiden (kryptischer Polymorphismus). Es ist eine Isolierung erst durch einen Gerinnungstest (Blutgruppenpolymorphismus) oder eine biochemische Analyse wie etwa Elektrophorese oder Gensequenzierung möglich. Einen Sonderfall stellt der chromosomale Polymorphismus dar. Hier unterscheiden sich die Individuen einer Population hinsichtlich ihrer Chromosomenzahl oder Chromosomenstruktur. Eine besondere Bedeutung hat der Inversionspolymorphismus bei vielen Arten erlangt. Er liegt dann vor, wenn innerhalb einer Population von den homologen Chromosomen zwei oder mehr Typen vorkommen, die als Umkehrungen (Inversionen) von Chromosomenabschnitten in Erscheinung treten. Hier handelt es sich nicht um Verschiedenheiten im genetischen Material, sondern um Verschiedenheiten in der Genreihenfolge im Chromosom. Eine Population kann polymorph oder monomorph für eine bestimmte Eigenschaft sein. Selbstverständlich kann die Wirkung eines Gens nur dann erkannt werden, wenn von ihm mehr als zwei verschiedene Allele vorhanden sind. Nehmen wir etwa an, wir könnten in einer Population direkt die Gene erfassen. Es lägen etwa von den Genen A, B, C und D nur für A und C verschiedene Allele vor (etwa A1/A2 und C1/C2), während B und D nur als Allel B1 bzw. D1 vorkämen. In der Folge betrachten wir einen beliebigen Genlocus mit zwei Allelen, die üblicherweise mit Großbuchstaben (A, S, … und Kleinbuchstaben (a, s, ..) bezeichnet werden. Das Hardy Weinberg-GesetzIm Jahr 1908 fanden der Mathematiker Godfrey Harold Hardy und der Arzt Wilhelm Weinberg unabhängig voneinander ein Gesetz, das die Verteilung von Erbmerkmalen in Populationen beschreibt. Wir betrachten eine Population mit Zufallspaarung, in der schwarze, graue und weiße Mäuse nebeneinander vorkommen. Die Eigenschaften schwarz und weiß werden von den Allelen eines Gens bestimmt. SS bedingt schwarze Färbung, ss weiße; die Heterozygoten Ss wären grau. Wenn wir den Genpool der Population betrachten (siehe Abb. 2), so finden wir, dass die beiden Allele S und s in der Population eine unterschiedliche Häufigkeit haben. Bezeichnen wir die relative Häufigkeit von S mit p und die von s mit q, so können wir, wenn wir Selektion ausschließen, die Population in Bezug auf diesen einen Genlocus durch die Angabe p=0,4 (40%) und q=0,6 (60%) beschreiben.
Ableitung des Hardy- Weinberg-Gesetzes. A: Die Elternpopulation besteht aus SS-, Ss- und ss-Individuen, die sich zufällig paaren. Die Wahrscheinlichkeit für jeden Paarungstyp ergibt sich aus den Häufigkeiten der beteiligten Genotypen. Die möglichen Nachkommen aus den einzelnen Paarungen sind in der Tabelle eingetragen. Durch Addition der Erwartungswerte der einzelnen Genotypen ergibt sich deren relative Häufigkeit unter den Nachkommen. B: Der Eltern-Genpool besteht aus den Allelen S und s. Diese kombinieren sich zufällig und bilden so die Individuen der Nachkommenschaft. Die Häufigkeit der Genotypen ergibt sich aus der Einzelhäufigkeit von S und s.
Wir nehmen an, dass alle Individuen einer Population gleiche Wahrscheinlichkeit haben, Nachkommen zu produzieren. Wir können daher die genetische Zusammensetzung der Nachkommen-Generation nach den Gesetzen der Wahrscheinlichkeit berechnen. Es gilt allgemein: Die Wahrscheinlichkeit, dass zwei voneinander unabhängige Ereignisse zugleich auftreten, ist gleich dem Produkt der Einzelwahrscheinlichkeiten. Es ist die Wahrscheinlichkeit, dass bei einem Würfel eine Eins geworfen wird, gleich 1/6. Die Wahrscheinlichkeit, dass bei gleichzeitigem Werfen von 2 Würfeln sowohl der eine als auch der andere eine Eins zeigt, ist gleich dem Produkt der Einzelwahrscheinlichkeiten, also 1/6 * 1/6 = 1/36. Mit Hilfe dieser Überlegung kann man das Gesetz über den genetischen Aufbau einer Mendel-Population ableiten. Dazu können wir sowohl von den Individuen der Population selbst ausgehen als auch vom Genpool. Im ersten Fall müssen wir uns zunächst fragen, wieviel verschiedene mögliche Paarungen es gibt und wie häufig jeder der einzelnen Paarungstypen zu erwarten ist. Da es sich um Zufallspaarung handelt, ist die Wahrscheinlichkeit, dass gerade etwa zwei schwarze Individuen zusammenkommen, gleich dem Produkt aus den Einzelhäufigkeiten. Wenn p die relative Häufigkeit der schwarzen, h die der grauen und q die der weißen Individuen ist, dann erwarten wir also die Kreuzung Schwarz x Schwarz mit der Häufigkeit p * p = p² und Grau x Weiß mit der Häufigkeit h * q + q * h usw. Nachdem wir so die Erwartungshäufigkeit für die verschiedenen Paarungen ermittelt haben, müssen wir uns noch überlegen, welche Nachkommen jeweils aus den Kreuzungen zu erwarten sind. Addieren wir nun die Erwartungshäufigkeiten der drei möglichen Farbtypen schwarz, grau oder weiß unter den Nachkommen, so erhalten wir die theoretische Verteilung. Es ergibt sich, dass Schwarz : Grau : Weiß in der Nachkommenschaft so verteilt sein wird wie (p + ½ h)² : 2(p + ½ h)(q + ½ h) : (q + ½ h)² Leichter kommt man zu einem Ergebnis, wenn wir vom Genpool ausgehen. In diesem hat das Allel S die Häufigkeit p und s die Häufigkeit q. In der nächsten Generation werden die Allele zufällig zu Zweiergruppen kombiniert, wie es bei der Verschmelzung der Gameten ja tatsächlich der Fall ist. Wir erhalten die Kombinationen SS, Ss und ss mit der Häufigkeitsverteilung Dieses Ergebnis entspricht dem Hardy-Weinberg-Gesetz. Es kann leicht bewiesen werden, dass die mit verschiedenen Methoden gefundenen Verteilungsproportionen einander entsprechen. Rechenbeispiele: Sind in einer Population die Allele A (Häufigkeit p = 0,5) und a (Häufigkeit q = 0,5) jeweils zu 50% vorhanden, so gilt: p + q = 1 (100%) 0,5 + 0,5 = 1 Die Genotypenhäufigkeiten erhält man durch das Hardy-Weinberg-Gesetz: p² + 2pq + q² = 1 0,25 + 0,5 + 0,25 = 1 (25% AA-Genotypen, 50% Aa-Genotypen, 25% aa-Genotypen) In der Regel findet man in der Natur einen Genotyp und rechnet daraus die anderen Genotypen aus. Nehmen wir an, wir finden, dass ein rezessiven Genotyp aa in einer Population mit einer Häufigkeit von 13% gefunden wird, dann erhält man: q² = 0,13 --> q = 0,3606 --> p = 1 – q = 0,6394 daraus ergeben sich alle drei Genotypen: p² (AA) = 0,4088 (41 %) 2pq (Aa) = 0,4612 (46 %) q² (aa) = 0,13 (13%) Seltene rezessive Erbkrankheiten: Es gibt autosomal rezessiv vererbte Krankheiten wie die Phenylketonurie (PKU). Es handelt sich um eine Stoffwechselerkrankung, die mit Hilfe eines Tests nach der Geburt erkannt werden kann. Die PKU-Häufigkeit liegt bei ungefähr 1:8000 bis 1:10.000 Neugeborenen. Die betroffenen Kinder können die Aminosäure Phenylalanin nicht abbauen, wodurch diese sich im Körper anreichert und Abfallprodukte erzeugt, die unbehandelt zu schweren geistigen Entwicklungsstörungen führen. Bestimmte Stoffwechselprodukte (Phenylketone), die mit dem Urin ausgeschieden werden, waren für die Erkrankung namensgebend. Wir nehmen an, dass PKU mit einer Häufigkeit von 1:10.000 auftritt. (Die Allelfrequenzen sind regional tatsächlich unterschiedlich zu beobachten.) q² (aa) = 0,000, das entspricht 1:10.000 Daraus errechnet sich q = 0,01 und p mit p = 1 – q = 0,99; Es ergeben sich nun alle drei Genotypen: AA-Typen sind phänotypisch gesund (sie haben nicht das "belastete" a-Allel; Aa-Typen sind phänotypisch gesund (sie sind "Überträger"); aa-Typen sind phänotypisch erkrankt (sie sind homozygot rezessiv); p² (AA) = 0,98 (98 %) Das heißt nichts anderes, als dass sich die Allele einer mit einer Häufigkeit von 1:10.000 rezessiv vererbten Krankheit zum überwiegenden Teil in Heterozygoten "verstecken". Eines von 10.000 neugeborenen Kindern ist von der Erbkrankheit betroffen, aber jedes fünfzigste Kind (2pq = ca. 0,02) ist Überträger. In einer Stadt mit 100.000 Einwohnern leben demnach zehn Erbkranke (aa), jedoch zweitausend Überträger. Dies gilt nur für eine einzige Erbkrankheit. Da es aber mehrere tausend rezessive Erbkrankheiten gibt (die genaue Zahl ist unbekannt), von denen die meisten sehr selten sind, gilt als statistisch gesichert, dass jeder Mensch Überträger (Aa) für mehrere rezessive Erbkrankheiten ist. Der Genetiker spricht von einer "genetischen Bürde" (Genetic load) (2). Das Hardy Weinberg-Gesetz kann mit jedem Taschenrechner leicht berechnet werden. Es ist auch möglich, die folgende Excel-Tabelle zu verwenden:
Abb. 3; Excel-Erscheinungsbild für P² = 0,25 (25%), 2pq = 0,5 (50%), q² = 0,25 (25%)
Abb.: 4; Excel-Erscheinungsbild für P² = 0,4675 (46,75%), 2pq = 0,4325 (43,25%), q² = 0,1 (10%) (4) Die Fitness (ein einfaches Selektionsmodell)"Die allermeisten Mutationen oder Genveränderungen wirken sich nachteilig aus. Über Selektionsprozesse pflanzen sich jedoch die Individuen mit den günstigeren Genen erfolgreicher fort, und negative Mutationen gehen wieder verloren." Diese oder ähnliche Sätze findet man im Internet zu Dutzenden. Der Fehler, der hier auftritt, liegt in der Missachtung (oder Unkenntnis) der Fitness, die bei sexueller Fortpflanzung zu beobachten ist. In der Folge wird hier nur ein sehr einfaches Ein Genlocus-2 Allel-Modell besprochen, um zu zeigen, was – auch von Biologen – allzu oft übersehen wird. Man muss sich vor Augen führen, dass das bisher besprochene Hardy-Weinberg-Gleichgewicht in der Natur nicht immer anzutreffen ist. Die Formel p² + 2pq + q² = 1 ist nur ein mathematischer Ausdruck für einen Idealfall, auf den man sich beziehen kann, wenn man die Verteilung von Realpopulationen betrachtet. Gemessene Abweichungen von diesem Gesetz deuten (fast) immer auf Selektionsprozesse hin. Es wäre allerdings völlig unsinnig das Hardy-Weinberg-Gesetz für unnütz zu halten. Es ist ein allgemein gültiges Gesetz, das uns einen guten Blick in die mathematisch-statistische Struktur von Mendel-Populationen gewährt. Nicht alle Individuen einer Population sind gleichartig. (Um ganz genau zu sein: Keines gleicht dem anderen aufs Haar. Neben morphologischen Unterschieden wie Größe, Gewicht oder Färbung finden wir auch solche in Bezug auf Überlebenswahrscheinlichkeit oder Fruchtbarkeit. Gerade diese Tatsache ist für die Eigenschaften der Genzusammensetzung wichtig. Fitness kann sich aus verschiedenen Komponenten zusammensetzen. (z.B.: Puppenschlüpfrate bei Insekten, Zahl der produzierten Eier, Langlebigkeit, Paarungserfolg, Entwicklungsgeschwindigkeit ,Körpermasse u.a.) Erwartet man z.B. aus der Kreuzung Aa x aa die beiden Genotypen mit gleicher Häufigkeit, so zeigt uns eine Abweichung von der Erwartung eine unterschiedliche Fitness der beiden Genotypen an. In der Populationsgenetik wird die "Wrightsche Fitness" mit W abgekürzt. Der (relative) Maximalwert wird mit 1 angenommen. Hat ein Genotyp die Fitness von W = 0, dann ist er entweder nicht lebensfähig oder stirbt einen genetischen Tod. Der Genotyp fällt für die Evolution aus. Beispiel: Wenn die Aa-Typen die Fitness 1 haben, ergibt sich die relative Fitness eines Genotyps (aa) aus: Waa = (Zahl aa)/(Zahl Aa) So wie ein bestimmter Genotyp eine bestimmte Fitness (W1) besitzt, kann auch die Gesamtfitness (Wges) einer Population berechnet werden. Sie entspricht dem arithmetischen Mittel aus der Individualfitness aller ihrer Individuen. Wenn man neben dem Ausdruck der Fitness (W) noch den Ausdruck des Selektionskoeffizienten oder Selektionsnachteils (s) einführt, so gelten folgende Definitionen: Vollständige Dominanz (nur die aa-Typen werden gegenselektiert): Unvollständige Dominanz (Aa und aa werden gegenselektiert: Überdominanz (auch Heterosis genannt, AA und aa werden gegenselektiert): s = 0 --> W = 1: Vollständige Fitness Wir berechnen nun den Selektionskoeffizienten: Wenn q0 die relative Häufigkeit des Allels a in der "Ausgangsgeneration" (die erste beobachtete Generation) ist, so ist q1 die relative Häufigkeit des Allels a in der Tochtergeneration. Streng genommen gilt diese Formel nur für sogenannte "diskrete" Generationen, wie sie z.B. bei einjährigen Pflanzen auftreten. In der Realität gibt es aber zwischen den Generationen keine scharfen Trennungslinien. Trotzdem ist der vorliegende Fehler zu gering, als dass er in Betracht gezogen werden müsste. Vorliegende Formel ist daher gültig. Kennt man durch Auszählung q0 und q1, so errechnet sich daraus der Selektionsfaktor s folgendermaßen: s = (q0 - q1)/(q²0(1 - q1)) Wir nehmen an, dass es in einer Tierpopulation verschiedene Genotypen gibt bezüglich Temperaturempfindlichkeit. Die Genotypen AA und Aa sind weitgehend kälteresistent, die aa-Genotypen sind es nicht (vollständige Dominanz). Die aa-Typen sind ursprünglich zu 50% vertreten (q²0 = 0,5). Es kommt zu einer Kältephase, was zur Folge hat, dass innerhalb einer Generation die aa-Typen auf 45% (q²1 = 0,45) abnehmen. Der Selektionskoeffizient s errechnet sich dann nach der obigen Formel: s = (0,707 – 0,671)/0,5(1 – 0,671) = 0,22 Es gibt verschiedene Gründe, diese Rechnung für ungenau zu halten. Auf diese Kritik sei im letzten Kapitel eingegangen. Kennt man den Selektionskoeffizienten, dann kann man die Allelfrequenzen der folgenden Generationen berechnen. q1 = (q0 (1 - sq0))/(1 - sq²0) Aus q ergeben sich mit Hilfe des Gesetzes von Hardy-Weinberg alle Allel- und Genotypfrequenzen. Beachte: W = 1 – s. W (Fitness) und s (Selektionskoeffizient) können nur Werte zwischen 0 und 1 annehmen. Das wurde im Excel-Modell Wir betrachten nun ein simples 1-Genlocus-2-Allel-Modell. Die Allele werden mit A (Häufigkeit p) und a (Häufigkeit q) bezeichnet. Dementsprechend existieren die Genotypfrequenzen AA (Häufigkeit p²), Aa (Häufigkeit 2pq) und aa (Häufigkeit q²).
Abb. 5.: Startfrequenzen qA = 0,99 (99%), qa = 0,01 (1%) WAA = 0,8 WAaAa = 1,0 Waa = 0,1 Wir haben es hier mit einem Heterozygotenvorteil (WAA hat den höchsten Fitnesswert) zu tun, der in tausenden Selektionsversuchen mit verschiedenen Drosophilaarten bestätigt wurden. Warum Heterozygote sehr oft die höchste Fitness haben, ist noch nicht vollständig geklärt. Es gibt verschiedene Theorien, die hier aus Platzgründen nicht erwähnt werden. Wir erkennen in Abb. 5 eine scheinbar paradoxe Situation. Obwohl die Aa-Typen den höchsten Fitnesswert besitzen, pendeln sie sich bei einer Häufigkeit von nur 30% (2pq = 0,3) ein. Das hängt damit zusammen, dass die aa-Typen besonders stark gegenselektiert werden.
Abb. 6.: Wir betrachten einen extremen Fall von Heterosis: Startfrequenzen qA = 0,9684 (97%), q = 0,0316 (3%)
Abb.: 7: p²AA = 0,9801 2pqAa = 0,0198 und q²aa = 0,0,0001 Wir haben hier (Abb. 7) eine Population mit 98% AA- und 1,98% Aa-Typen. Sie haben die Fitness W = 1. Dieses Phänomen der Heterosis ist ein besonders wichtiger Evolutionsfaktor und wird von Kritikern (in der Regel ID-Vertreter und Kreationisten) übersehen. Es gibt zahlreiche Gründe dafür, dass Letal- oder Subletalallele trotz Selektion nicht aus einer Population verschwinden. Diese Gründe werden in der populationsgenetischen Fachliteratur ausgiebig beschrieben. (5) Zusammenfassung und Kritik( Die Angabe über die Zahl der Genloci beim Menschen variiert. Wenn wir die Zahl 20.000 annehmen, wobei 5% heterozygot (mit mindestens zwei Allelen) vorliegen, dann haben wir mindestens 1.000 Genloci mit jeweils mehreren Allelen. Wahrscheinlich ist die Zahl höher, weil noch nicht alle heterozygoten Genloci gefunden wurden. Die Zahl der möglichen verschiedenen Genotypen ist somit alleine beim Menschen astronomisch hoch. Bei 1000 Genloci mit jeweils 2 Allelen haben wir es mit einer irreal hohen Zahl (10300) von Kombinationsmöglichkeiten zu tun. ( ( A a (Cis-Typ) A a (Trans-Typ) | | | | B b b B In beiden Fällen haben wir es mit den doppelt heterozygoten Genotypen Aa/Bb zu tun, aber die relativen Positionen auf dem Chromosom führen zu unterschiedlicher Selektion. Evolutionsbiologisch verhalten sich beide Chromosomen nicht gleich (4). ( ( Aus dem Ektoderm hat sich zunächst ein Rohr abgespalten, was heute während der Embryonalentwicklung aller Wirbeltiere zu beobachten ist. Dieses Rohr wurde später zum Zentralnervensystem. Die Mutationen, die zur Wirbelsäule und zu unserem Zentralnervensystem führten, waren im homozygoten Zustand zunächst wahrscheinlich neutral. Aber auch dann, wenn sie gegenselektiert wurden, blieben sie (wie oben gezeigt) in den damaligen Populationen wegen der Heterosis erhalten. Irgendwann haben sich die Mutationen dann doch als vorteilhaft erwiesen, was zur Bildung der Wirbeltiere und ihrem Siegeszug führte. Mutationen müssen also nicht sofort homozygot "brauchbar" sein. Sie können das auch nach tausenden Generationen werden. Man benötigt keine komplexen Annahmen oder gar die These einer Unwahrscheinlichkeit, um die Entstehung neuer Tier- und Pflanzenstämme zu verstehen. ( Ein gravierender Fehler wird gerne übersehen. Es ist das Übergehen der Evolutionsgenetik. Als pars pro toto sei das Buch "Creatio" (5) erwähnt. Das Buch enthält über weite Strecken gesicherte Tatsachen (genetischer Code, Mendelsche Erbgesetze, Plattentektonik usw.). Diese Details findet man auch in der seriösen Fachliteratur und in populärwissenschaftlichen Werken. Große Teile des Buchs betreffen die "Makroevolution" als Lieblingsthema der Kreationisten. Im Kapitel "Molekularbiologie und Genetik" heißt es auf Seite 165:
Das Argument: "Makroevolution kann man nicht beobachten, weil sie viel zu langsam stattfindet, scheint sehr einleuchtend zu sein. Dabei wird aber von höheren Tieren und Pflanzen und deren Generationszeiten ausgegangen. Denkt man an Bakterien, die sich unter optimalen Bedingungen alle 15 Minuten teilen können, so kann man an ihnen in etwa 3 Jahren die Veränderungen beobachten, die in über 100.000 Generationen erfolgen. Diese Anzahl soll nach der Meinung vieler Wissenschaftler größenordnungsmäßig ausreichen, um in höheren Lebewesen aus einem einfachen Vorläufer (Grubenauge) unser heutiges Linsenauge zu entwickeln. Die Experimente mit dem "Haustier der Genetiker", der Fruchtfliege Drosophila, brachten nie etwas anderes als geschädigte Fruchtfliegen hervor. Man sucht im Buch vergeblich nach Begriffen wie "haploid", "diploid", "Heterosis", "Evolutionsgenetik" usw. Bei der Evolutionsgenetik handelt es sich immerhin um eine Disziplin der Biologie, die bereits viele Antworten und Erklärungen anbieten kann. Stattdessen werden Behauptungen aufgestellt, die keiner Überprüfung standhalten. So wird als "Beweis" für die Unmöglichkeit der "Makroevolution" folgendes Argument angeboten: "Neues Erbmaterial, d. h. neue, funktionsfähige Gene und neue Strukturen, also neue Proteine, müssten durch Mutationen zustande kommen können. Dieser Nachweis konnte bisher nicht erbracht werden." Tatsächlich wurden an der Wiege der Chorda- und Wirbeltiere (also alles vom Hai über die Eidechse bis zum Menschen) nur zwei Genverdoppelungen benötigt, um zwei Rohre zu erzeugen. Die entsprechenden Gene wurden zu Beginn aufgrund bekannter Heterosis-Mechanismen in die Population eingebaut, unabhängig davon, ob es sich um positive oder negative Mutationen handelte. Mit anderen Worten: Neues ist in der Evolution meist weder nützlich noch tödlich, es war oder ist einfach da. Später hat die Evolution durch viele kleine Schritte weiter Neues auf Neues geschichtet. Was die Kreationisten als "Makroevolution" bezeichnen, ist nichts anderes, als eine ursprünglich eher simple Veränderung, die so lange als genetische Reserve in der Population blieb, bis ihre Fitnesswerte – aus welchen Gründen auch immer – anwuchsen. Literatur:(1) R. Lewontin: "The Genetic Basis of Evolutionary Change", Columbia University Press. (2) B. Wallace: "Die genetische Bürde", Gustav Fischer Verlag (Orig.: "Genetic Load", Prentice Hall) (3) D. Sperlich: "Populationsgenetik", Gustav Fischer Verlag (4) Rudolf Öller: "Über die Effekte von Selektion, Rekombination und Gen-interaktion an Allelen von gekoppelten Enzymloci und einem balancierten Letalsystem in Modellpopulationen von Drosophila melanogaster." (Dissertation an der Universität Tübingen, 1981) (5) Alexander vom Stein: "Creatio" (Daniel Verlag) |
|
|
Vorarlberger Bildungsserver |
| Anatomie/Physiologie | Botanik | Cytologie | Evolution | |
| Genetik | Humanbiologie | Ökologie | Sexualbiologie | Zoologie |
| Geschichte | Texte, Referate | Sehenswert | Kontakt | Physik |
Zwei Genetikthriller vom Autor dieser Seite |
.png)
Rudolf Oeller:Typhons RacheThriller über die vernichtende Kraft der Rache und den Traum vom ewigen Leben im Diesseits.
|
.png)
Rudolf Oeller:Typhon DistrictThriller über eine Gruppe junger Wissenschaftler, die an ihrer Gier zugrunde ging.
|
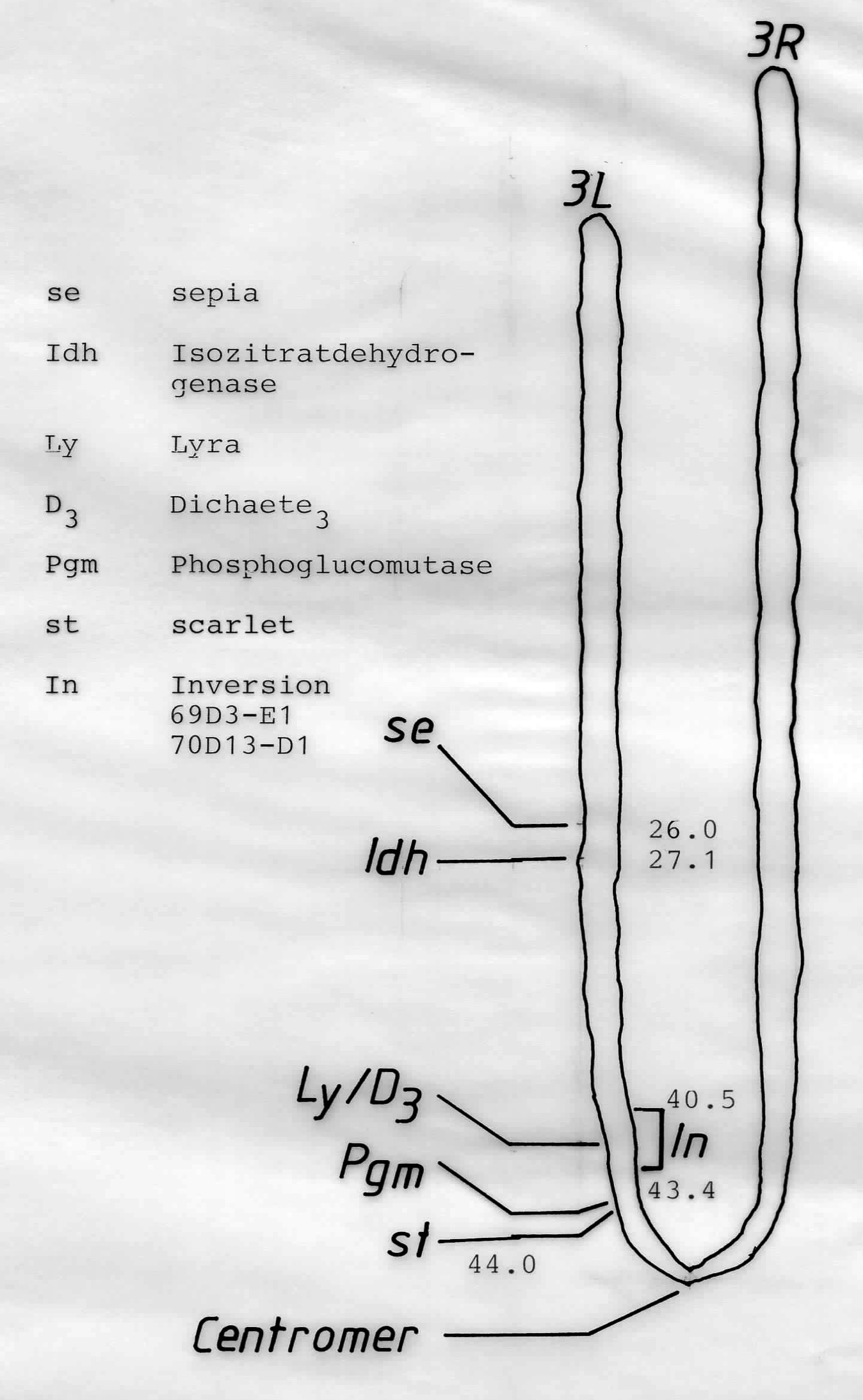
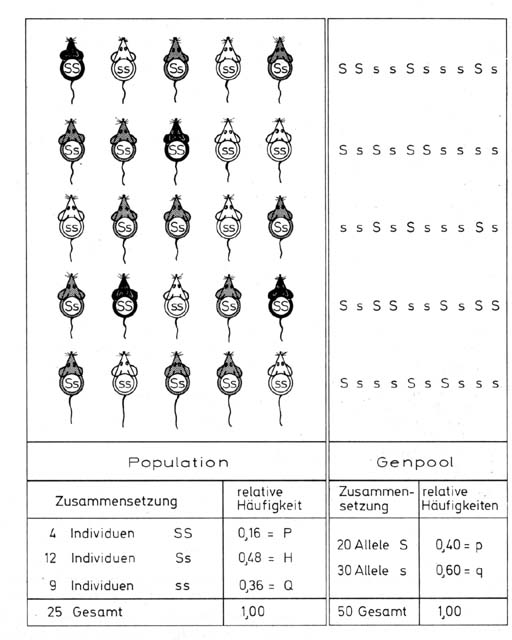 Abb. 2:
Abb. 2: .png)
.png)
.png)
.png)
.png)
 Es gibt noch eine Reihe weiterer Beispiele für Mutationen, die sich nachgewiesenermaßen vorteilhaft für ihre Träger auswirken. Die meisten davon beruhen aber auf dem Verlust oder der Beschädigung einer bestehenden Struktur. Damit Makroevolution stattfinden kann, reicht es nicht aus, dass sich die Existenz positiver Mutationen nachweisen lässt. Ob eine Mutation positiv, also vorteilhaft ist, hängt von den Lebensbedingungen (Umwelteinflüssen, Selektionsfaktoren) ab. Um Makroevolution durch eine Anhäufung von Mutationen plausibel zu machen, müsste man zeigen können, dass auf diesem Weg etwas qualitativ Neues entstehen kann. Neues Erbmaterial, d. h. neue, funktionsfähige Gene und neue Strukturen, also neue Proteine, müssten durch Mutationen zustande kommen können. Dieser Nachweis konnte bisher nicht erbracht werden...
Es gibt noch eine Reihe weiterer Beispiele für Mutationen, die sich nachgewiesenermaßen vorteilhaft für ihre Träger auswirken. Die meisten davon beruhen aber auf dem Verlust oder der Beschädigung einer bestehenden Struktur. Damit Makroevolution stattfinden kann, reicht es nicht aus, dass sich die Existenz positiver Mutationen nachweisen lässt. Ob eine Mutation positiv, also vorteilhaft ist, hängt von den Lebensbedingungen (Umwelteinflüssen, Selektionsfaktoren) ab. Um Makroevolution durch eine Anhäufung von Mutationen plausibel zu machen, müsste man zeigen können, dass auf diesem Weg etwas qualitativ Neues entstehen kann. Neues Erbmaterial, d. h. neue, funktionsfähige Gene und neue Strukturen, also neue Proteine, müssten durch Mutationen zustande kommen können. Dieser Nachweis konnte bisher nicht erbracht werden...